CIENCIAS BIOLÓGICAS Y DE LA SALUD
Una nueva herramienta para la edición génica en bacterias Gram-negativas
El trabajo colaborativo entre investigadores del CONICET y de la Universidad Tecnológica de Dinamarca derivó en el desarrollo de un método que minimiza los costos, aumenta la eficiencia y reduce los tiempos para la manipulación genética de microorganismos.
{kind=link}
Investigadores del CONICET participaron en la construcción de una serie de herramientas de ingeniería genética basadas en CRISPR/Cas9 para facilitar la manipulación del ADN de bacterias Gram negativas (caracterizadas por presentar doble membrana celular) y acelerar trabajos de investigación básica y aplicada. El método presentado permite la edición de múltiples genes en simultáneo, reduciendo los costos. El trabajo, publicado en la revista Nature Communications, fue realizado por Román Martino y Andrea Smania, del Centro de Investigaciones en Química Biológica de Córdoba (CIQUIBIC, CONICET-UNC) en colaboración con el grupo “Microbiología Ambiental de Sistemas”, del Novo Nordisk Foundation Center for Biosustainability (Universidad Técnica de Dinamarca).
“En la actualidad, las herramientas de ingeniería genética con las que contamos para la edición genómica en microorganismos son lentas y tediosas. Esto ralentiza los proyectos de investigación, especialmente cuando el resultado depende de la modificación de más de un solo gen. La construcción de un rasgo o fenotipo complejo que requiere la meditación de, por ejemplo, 10 genes, toma varios meses”, explica Martino. El método de edición de bases múltiple presentado por los autores, basado en el procesamiento del ARN por la endoribonucleasa Cas6f, permite a los investigadores editar más de 10 genes simultáneamente en un único paso sin comprometer la eficiencia del proceso.
La construcción del vector pMBEC, que supone el punto de partida para este tipo de modificaciones, es económica y rápida gracias a un protocolo de pocos pasos que automatiza el diseño de los oligonucléotidos y se basa en el clonado por Golden Gate, un método que emplea solo una enzima de restricción. “Minimizar los costos, aumentar la eficiencia y reducir los tiempos de clonado son las principales ventajas de este desarrollo que hace accesible la tecnología a todos los grupos de investigación”, afirma Smania.
¿Qué implica este avance metodológico para la investigación genómica?
El desarrollo de CRISPR/Cas9 como herramienta para la edición génica ha generado una verdadera revolución en la manera en que se llevan a cabo modificaciones en el genoma de distintas especies, inaugurando una nueva era de la ingeniería genética. Esta tecnología se ha aplicado exitosamente en distintos organismos para insertar, reemplazar, modificar y/o eliminar genes. Cuando CRISPR/Cas9 se combina con una enzima (la deaminasa APOBEC1) que permite el cambio específico de una base citosina por timina, se constituye en lo que se conoce como editor de bases y posibilita generar modificaciones al nivel de nucleótidos individuales. Sin embargo, la científica señala que: “a pesar de los avances significativos que se han realizado en la última década, aún existen desafíos por superar para la implementación de esta tecnología, en especial para el estudio de bacterias no convencionales tales como el Pseudomonas putida, un microorganismo de interés industrial, o Pseudomonas aeruginosa, un patógeno de gran relevancia a nivel clínico”.
La innovación del método presentado por los autores se basa, en primer lugar, en la adaptación de un editor de bases para obtener una eficiencia de edición mayor al 90%. Posteriormente, con la incorporación de la endoribonucleasa Cas6f para el procesamiento del ARN, se amplía el sistema para la edición simultánea de más de 10 genes con una eficiencia mayor al 85%. Mediante la implementación de un protocolo simplificado para el diseño de los ARN guías y su ensamblado, es posible utilizar este sistema tanto para aplicaciones biotecnológicas como en estudios de la biología fundamental de bacterias, ampliando el uso y versatilidad de la tecnología CRISPR/Cas9.
¿Qué es un editor de bases?
“Un editor de bases está compuesto por dos enzimas: Cas9 y APOBEC1. Explicado en pasos simples, primero Cas9 reconoce y se une a un ARN, que la guía a la región del genoma que se busca editar. Una vez localizada, Cas9 produce el corte y abre la doble hebra de ADN, volviendo así accesible las citosinas que van a ser modificadas por timinas por la APOBEC1 (Fig. 1). Este cambio en las letras del ADN puede traducirse en la interrupción, la alteración o modificación de los niveles en los cuales se expresa una determinada proteína, editando consecuentemente las funciones celulares”, precisa Smania.
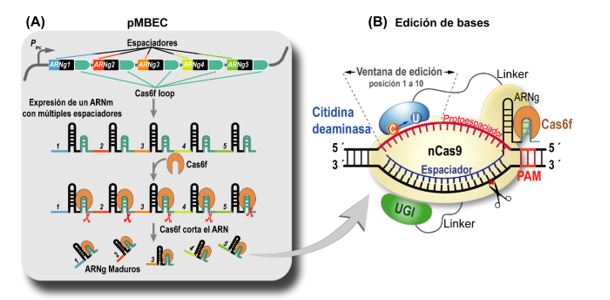
Fig. 1. El editor bases descrito en nuestro trabajo. (A) El ARN guía (ARNg) se expresa bajo un solo promotor y es procesado posteriormente por Cas6f para liberar las ARNg individuales. Esto permite que se editen simultáneamente lugares distantes en el genoma. (B) Un Cas9 deficiente (nCas9) es guiado por un único ARN guía (ARNg) a la secuencia protoespaciadora. La citidina desaminasa APOBEC1, fusionada con la nCas9, convierte la citosina en uracilo dentro de una ventana de edición; la dUTP se transforma posteriormente en dTTP durante la replicación del ADN. Esta estrategia se puede aplicar para uno o múltiples genes simultáneamente y así lograr la construcción de fenotipos bacterianos específicos.
Referencia bibliográfica:
Volke, D.C.*, Martino, R.A.*, Kozaeva, E., Smania, A.M., Nikel, P.I.. (2022). Modular (de)construction of complex bacterial phenotypes by CRISPR/nCas9-assisted, multiplex cytidine base-editing. Nature Communications 13, 3026. https://doi.org/10.1038/s41467-022-30780-z *equal contribution.